Usercases
Helicobacter pylori A45 total methylome analysis
In this example, we will use Snapper for the total methylome analysis of Helicobacter pylori A45 strain.
The running command:
(snapper) $ snapper -sample_fast5dir A45_multi -control_fast5dir A45_wga_multi -reference assembly_A45.fna -threads 16 -target_chr NZ_CP053256.1 -max_motifs 30 -outdir Results_A45
Here we set -max_motifs parameter to be 30 since H. pylori is known to have a huge number of different R-M systems (up to 30).
In addition, we specified -target_chr parameter to analyze only the main chromosome.
The first stage of the pipeline is raw signals collecting. It might take up to one hour depending on the dataset size. On average, a dataset consisting of 40-50 multifast5 batches is processed not longer than in 15-20 minutes.
Sample data collecting...
Batch 1 out of 40...
Batch 2 out of 40...
# and so on...
Control data collecting...
Batch 1 out of 40...
Batch 2 out of 40...
# and so on...
Next, the algorithm performs the Kolmogorov-Smirnov test to compare signal distributions for each 11-mer presented in the reference genome.
Forward strand signals processing...
Getting difsignals...
NZ_CP053256.1: 578090 out of 912990...
...
Just after all 11-mers that have significant signal shift have been extracted, they are written to the passed_motifs_{}.fasta file.
The next stage is a greedy motif extraction. In the console output, we can see the motif variant extracted on each algorithm iteration. For each motif, the algorithm checks if this motif should be merged with some of the motifs found on the previous iterations, which also can bee seen in the console output:
ITERATION 1 (55714 unexplained 11mers):
OBSERVING MOTIFS WITH LENGTH OF 4
OBSERVING MOTIFS WITH LENGTH OF 5
OBSERVING MOTIFS WITH LENGTH OF 6
Motif adjustment...
Motif adjustment...
(32929.810579835554, ('N', 'C', 'A', 'T', 'G', 'N'), (2, 3, 4, 5, 6, 7))
# the first extracted motif is NCATGN located from 2nd to 7th position in the 11-mer context
['NCATGN']
# just before the next iteration, the current set of potential methylation motifs is shown
ITERATION 2 (51145 unexplained 11mers):
OBSERVING MOTIFS WITH LENGTH OF 4
OBSERVING MOTIFS WITH LENGTH OF 5
OBSERVING MOTIFS WITH LENGTH OF 6
Motif adjustment...
Motif adjustment...
(27150.172723009397, ('N', 'G', 'C', 'G', 'C', 'N'), (2, 3, 4, 5, 6, 7))
['NCATGN', 'NGCGCN']
# one more motif was added to the methylation motifs set
ITERATION 3 (47632 unexplained 11mers):
OBSERVING MOTIFS WITH LENGTH OF 4
OBSERVING MOTIFS WITH LENGTH OF 5
OBSERVING MOTIFS WITH LENGTH OF 6
Motif adjustment...
Motif adjustment...
(25455.630042353463, ('N', 'C', 'A', 'T', 'G', 'N'), (3, 4, 5, 6, 7, 8))
(25455.630042353463, ('N', 'C', 'A', 'T', 'G', 'N'), (3, 4, 5, 6, 7, 8)) already has a supermotif!
Changed to (None, ('N', 'C', 'A', 'T', 'G', 'N'), (3, 4, 5, 6, 7, 8))
# here, we can see that a new extracted motif NCATGC is a duplicate,
# or, more generally, has a supermotif in the current motifs set. The algorithm modifies this motif according to the
# corresponding supermotif, extracts it, and goes to the next iteration
['NCATGN', 'NGCGCN']
# here, the motifs set has not been changed
ITERATION 4 (44097 unexplained 11mers):
...
When the algorithm extracts the desired number of motifs (in our case it equals 30) or reaches the limit of confidence level (in our case we used the default value equals 1000), it stops. The results include the generated list of potential methylation sites, where for each motif a corresponding confidence level and signal shift size are calculated. The table below shows the results returned by Snapper for H. pylori A45.
MOTIF |
conflevel |
effsize |
|---|---|---|
NCATGN |
32951.2 |
1.59 |
NGCGCN |
32109.6 |
1.11 |
NTGCAN |
19535.3 |
1.00 |
NGAACN |
21296.2 |
0.79 |
NGGCCN |
22838.8 |
1.42 |
NGATCN |
24323.8 |
0.46 |
NCCAGN |
22631.8 |
0.72 |
NCCATCN |
29952.0 |
1.54 |
NGAHTCN |
25034.2 |
0.72 |
NGGGGAN |
14548.9 |
0.58 |
ATTAATN |
16256.6 |
1.26 |
TCNNGAN |
16322.6 |
0.63 |
NTCNGAN |
18074.7 |
0.63 |
NGANTC |
11994.4 |
0.37 |
NGGAGAN |
11128.4 |
0.60 |
GTNNACN |
9591.0 |
0.92 |
NTCGAN |
4558.2 |
0.62 |
Generally, motifs with confidence level higher than 5000 might be used without additional verification regardless effect size. Thus, the NGATCN motif (actually just GATC) has a moderate signal shift (effsize < 0.5) but extremely high confidence level that indicates about motif correctness. In contrast, the NTCGAN motif has quite low confidence level but high effect size greater than 0.5, that seems satisfactory for motif inference.
Let’s consider some graphical output provided by the tool:
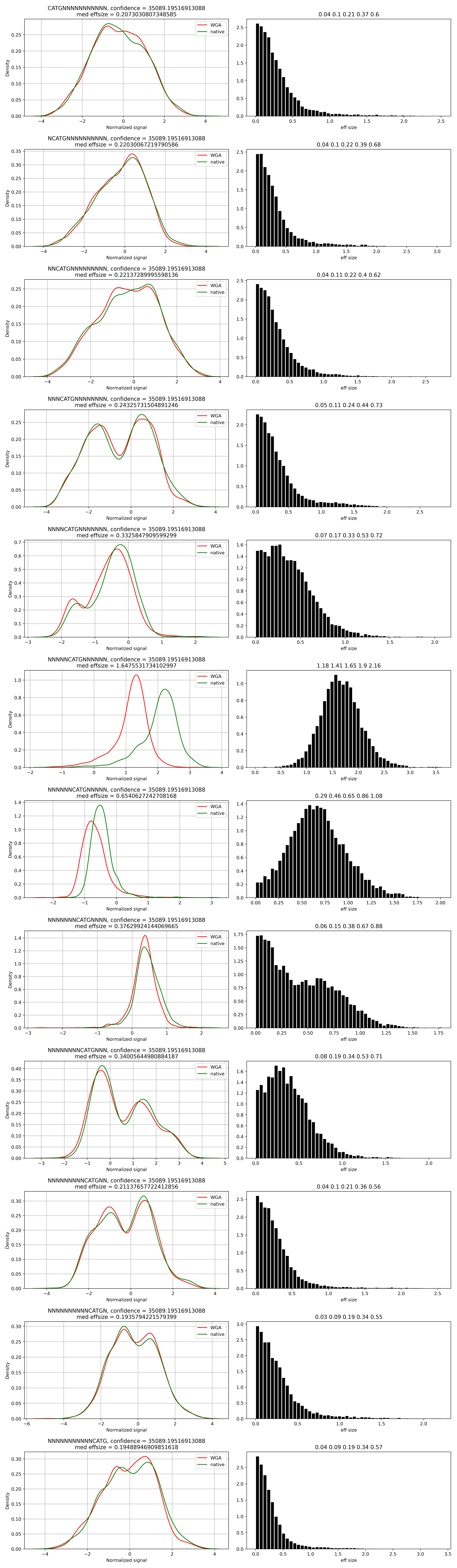
The left column shows the signal shift for all k-mers containing a considered motif (in our case CATG) in a different positions. The right columns shows the distribution of effect sizes collected from all k-mers represented in the considered genome and match the template on the left plot. If the motif is complete, this distribution should have one visible mode different from zero. Here, we can see a clear signal shift and unimodal distribution of effect sizes for NNNNNCATGNNNNNN variant that means that CATG is a complete methylation motif.
Numbers in the title of left distributions shows 10, 25, 50, 75, and 90 percentiles of the disctribution.
Long motifs inference
Snapper is designed mainly to extract short methylation motifs but has an additional module aimed to infer long motifs. When some motif seems incomplete, the algorithm tries to extend the motif enrichment heuristics and consider this motif as a part of a long motif.
Let’s consider Neisseria gonorrhoeae dataset analyzed with Snapper. The resulting list of motifs looks as follows:
>MOTIF_1 conflevel=93561.62457000425
NGGCCN
>MOTIF_2 conflevel=38224.7729351188
NGGNNCCN
>MOTIF_3 conflevel=35888.776173250444
NGCCGGCN
>MOTIF_4 conflevel=42282.94018790629
NTCACCN
>MOTIF_5 conflevel=31539.07710131262
NGGTGAN
>MOTIF_6 conflevel=8479.122038360483
NAGCGCCN
>MOTIF_7 conflevel=8042.143082793102
NGGCGCTN
>MOTIF_8 conflevel=8065.372900388755
NCCGCGGN
>MOTIF_9 conflevel=2327.5509766500168
NAGCGCTN
>MOTIF_10 conflevel=1545.3452789244398
NGCAN
During the analysis, Snapper identified NGCAN and NGGTGAN motifs as probably incomplete. In such cases, the algorithm tries to consider these motifs
as TRD1 binding sites and performs an additional local motif enrichment in order to find potential TRD2 sequence. The results are written to motif_refine folder,
where long_contexts.fasta file contains all genome contexts that seem to bring modification and contain the considered motif, and long_motif_variants.tsv file
contains the most over-represented long motif variants.
Let’s observe these results for NGCAN:
GCANNNNNNNNTGC 441265.7331442257
GCANNNNNNNNTG 237450.2481013803
GCANNNNNNNNTNC 214668.52967371218
GCANNNNNNNNTGCNG 174776.86134567283
GCANNNNNNNNTGCC 155828.7937985875
GCANNNNNNNNTGCNNC 146477.99146127273
GCANNNNNNNCTGC 137146.54147585746
GCANNNNNNNTTGC 135734.2313062486
GCANNNNNNGNTGC 123040.0689565634
GCANNNNNNTNTGC 121585.02483750567
GCANNNNNNNNTGCG 118720.66862643362
GCANNNNNCNNTGC 118187.48054433338
GCANNNNNTNNTGC 116974.78151809816
GCANNNNNNNNTGNNG 112591.28596115313
GCANNNNNNNNTGCNNG 112378.98428738883
GCANNNNNNCNTGC 110827.19675016444
GCANNNNNNNNTGCA 108986.77057724852
GCANNNNNGNNTGC 106590.62486627264
GCANNNNNNNNTGCNNT 100837.39703402991
GCANNNNNNNNTGNC 100265.40043701489
Again, here we have a number of variants and corresponding confidence levels (which are formally just chi-square statistics values). The GCANNNNNNNNTGC variant has the highest confidence level, but a more important thing that all other variants are quite close to it and generally describe the same pattern with some small deviations. So, the top variant being a consensus of this motif list is an actual long methylation site in N. gonorrhoeae.
Now, let’s check the NGGTGAN results:
GGTGANNNNNNNGNNG 143671.66760353974
GGTGANNNNNNCG 119845.58600206679
GGTGANNNNNNNGC 107939.88739540703
GGTGANNNNNNTG 105261.78657207903
GGTGANNNNNTNG 104221.77196083494
GGTGANNNNNNNNTNG 104084.1138691476
GGTGANNNNNNNGNT 104009.9046302189
GGTGANNNNNNNNTT 95939.67443296607
GGTGANNNNNNNNCNG 93002.75191853297
GGTGANNNNNNNGNC 88002.94759049421
GGTGANNNNNNNTT 87275.13589750837
GGTGANNNNNNNNAA 82825.6859630761
GGTGANNNNNTT 80897.32351811985
GGTGANNNNNTNNT 79271.31738543115
GGTGANNNNNNNTNNG 78790.29620219412
GGTGANNNNNGNNT 77130.00092316889
GGTGANNNNNNNNTC 76254.08160217352
GGTGANNNNNNTNNC 75652.85020331324
GGTGANNNNNNNGT 75652.85020331324
GGTGANNNNNNNNGC 74920.64477911562
Okey, we have quite high confidence value for the top variant. However, TRD2 sequence variants here are not similar to each other. The absence of a clear TRD2 consensus indicates that we should not consider NGGTGAN as a part of a long motif. Actually, it is a short individual motif in N. gonorrhoeae.
The results curation
In general, Snapper works well in fully-automated mode but sometimes it can be too sensitive. We intentionally set the default confidence level to be very low so that the algorithm could catch modifed contexts even with low coverage. Here, we will consider Thermacetogenium phaeum DSM 12270. The results we have obtained with Snapper are following:
>MOTIF_1 conflevel=98330.21053706403
NCCTCCN
>MOTIF_2 conflevel=88903.54075233328
NGGNCCN
>MOTIF_3 conflevel=107898.36547102229
NGATCN
>MOTIF_4 conflevel=142374.31120032488
NCGCGN
>MOTIF_5 conflevel=62056.54673082095
RAACTCN
>MOTIF_6 conflevel=55031.763919100005
NCTACTN
>MOTIF_7 conflevel=33127.41949287399
NTGGCCAN
>MOTIF_8 conflevel=25334.307196869693
NCAGAAAN
>MOTIF_9 conflevel=21550.144654935317
NCCCAAGN
>MOTIF_10 conflevel=17494.734513775027
NCCCGAGN
>MOTIF_11 conflevel=5564.894624926862
NCCAGN
>MOTIF_12 conflevel=369.22147225776064
NANGGTN
>MOTIF_13 conflevel=340.2454317151567
NCNGGTTN
>MOTIF_14 conflevel=332.5902949742641
NGGATC
>MOTIF_15 conflevel=277.9235875118016
NGGNCN
>MOTIF_16 conflevel=294.35993058820924
NATAN
>MOTIF_17 conflevel=281.9139853540567
NCNTCN
>MOTIF_18 conflevel=167.77079569128034
NGANTN
>MOTIF_19 conflevel=179.4513513422786
NTNTCN
>MOTIF_20 conflevel=160.10321003660576
NGGTGCN
Here, NCCTCCN, NGGNCCN, NGATCN, NCGCGN, RAACTCN, NCTACTN, NTGGCCAN, NCAGAAAN, NCCCAAGN, NCCCGAGN, NCCAGN motifs have a satisfactory confidence level. However, we can see a lot of sequences with confidence level lower than 500. If we consider graphical outputs for most these motifs we will not see a significant signal shifts but there is one more thing that helps to ensure that they are not actual motifs. According to REBASE, short methylation motifs 1) do not contain individual non-degenerate bases surrounded by ‘N’ and 2) should contain at least 4 non-‘N’ bases. Motifs NANGGTN, NCNGGTTN, NGGNCN, NCNTCN, NGANTN, NTNTCN do not satisfy the first rule, while NATAN do not satify the second rule so all these motifs should be removed. NGGATC is an occasionally appeared submotif for GATC and should be removed too. GGTGC motif has the lowest confidence level but a stable signal shift in the NNNNNNGGTGCNNNN varant.
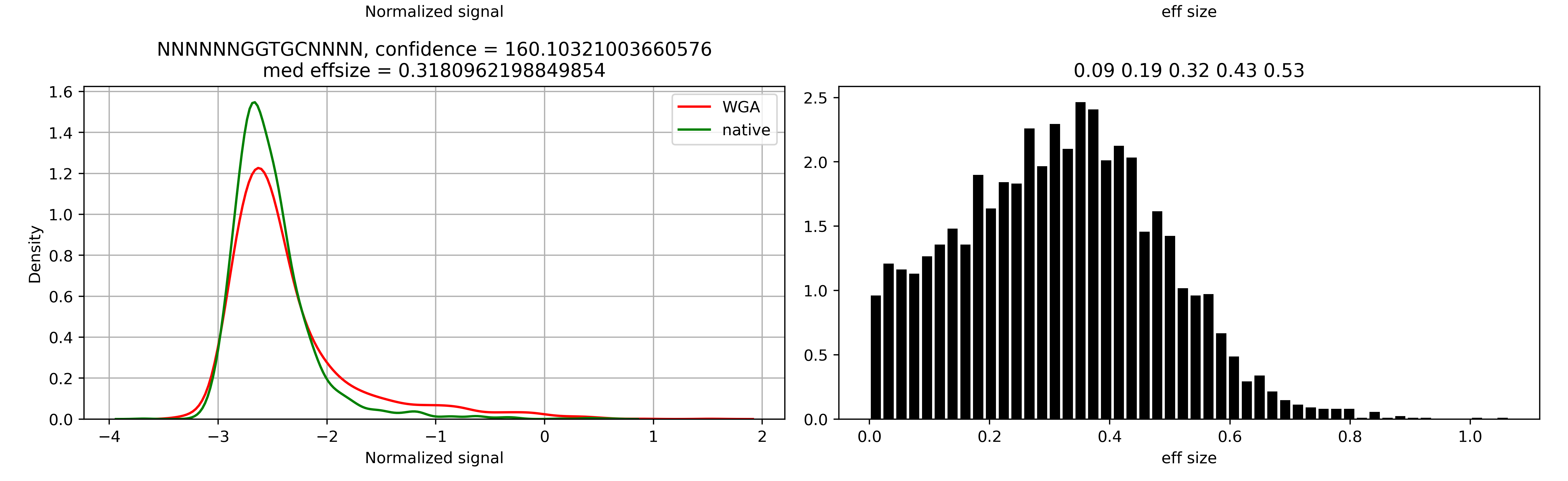
Indeed, the effect size value is quite good here, but the distributions for control and native samples have the same mode, and all the difference is caused by a longer “right tail” in the WGA signal distribution. Thus, we can conclude that GGTGC is not an individual methylation motif.
Main points
Here, we summurize some motif inference recommendations:
Generally, if the confidence level of a considered motif is greater than 5000, it is most probably an individual motif regardless signal shift size.
Generally, if the signal shift (effsize) of a considered motif is greater than 0.5, it is most probably an individual motif.
If some motif has a confidence level lower than 5000 and a signal shift lower than 0.5, it should be manually verified via corresponding signal distributions plots observation. If sample and control distributions have a common mode (one ore more), it usually indicates that motif sequence is incomplete.
If some motif has a confidence level lower than 500 and a signal shift lower than 0.25, it is most likely not to be an individual motif.
Demo-dataset
The demo-dataset is available on our FTP: http://download.ripcm.com/snapper_test/.
The demo-dataset includes multi-fast5 files for native H. pylori A45 strain (control folder) and its hpy mutant disrupted in the gene encoding CATG-specific methyltransferase (mutant folder).
The reference genome is available on GenBank: GCF_000333835.2.